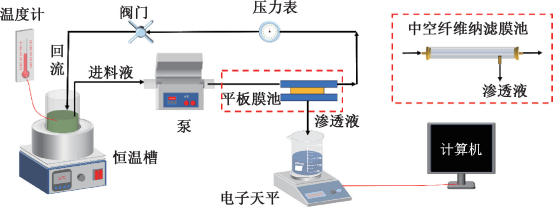
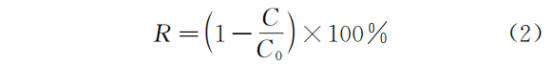清华大学孙文俊团队ES&T:纳米碳介导的过氧乙酸均解引起的连续电子转移产生预水合电子促进PFOA降解
图片摘要

成果简介
近日,清华大学环境学院薛雅内助理研究员和孙文俊副研究员团队在环境领域著名学术期刊Environmental Science & Technology上发表了题为“Prehydrated Electrons Activated by Continuous Electron Transfer Stemmed from Peracetic Acid Homolysis Mediated by Diamond Surface Defects for Enhanced PFOA Destruction”的研究论文。本文确定了过氧乙酸(PAA)基深度水处理技术(AWT)中连续电子转移的非自由基途径,该途径偏离了已报道的单线态氧、单电子或双电子转移路径,并探索了由该途径引发的强还原性预水合电子对脱除全氟化合物的关键作用。本研究构建的PAA/金刚石(NDs)体系在特定水质条件下可大幅减少AWT对催化剂和化学品的需求。
引言
深度水处理技术(AWT)旨在产生可降解水中新污染物的侵略性氧化或还原活性物质。这些化学过程包括以过氧化物为基础的AWT(例如,过氧单硫酸盐,PMS;peroxydisulfate PDS;过氧乙酸,PAA;过氧化氢(H2O2),通过相对弱的O-O键(一般键能为144 kJ/mol~213kJ/mol,S-S键能为266 kJ/mol, H-O为463 kJ/mol, C-C为332 kJ/mol, C-O为326 kJ/mol)均裂产生氧化自由基。PAA(CH3CH2COOH)最初作为氯消毒替代品,已被证明通过类似的活化方法(即紫外线、金属基催化剂和碳基催化剂)产生各种强氧化自由基。此外,PAA的pKa(8.2)高于氯(pKa=7.5),亨利定律常数(4.68×102M atm-1<7.36 ×104M atm-1),表明PAA的pH应用范围更广,且在水处理过程中的溶解度更佳。PAA-AWT处理水中新污染物的研究也因此逐步受到关注。在纳米碳(NC)-过氧化物的AWT系统中,NC表面的多介质配位机制主要包括三个方面:1)活性物质(RS)途径,NC失电子过氧化物得电子,电子转移路径(ETP)是从NC到过氧化物。2)非RS途径(电子还原),是指通过电子转移进行过氧化物氧化,ETP从污染物到NC/自由基再到过氧化物。3)非RS途径(单线态氧,1O2),过氧化物失电子并产生自由基,自由基的自反应产生1O2。这里ETP是从过氧化物到NC。然而,以上途径仅限于单步电子反应,当前对多通道或连续电子转移路径尚未取得探索结果。此外,在目前对非均相PAA-AWT反应机制的研究中,O-OH和/或O-H裂解产生的RS仍然是主要的反应物质,也有人提出通过其他途径,包括持久自由基和单电子或双电子转移过程来解离PAA,但关于PAA-AWT过程中电子转移引发的非RS通路的报道有限。
鉴于饮用水源和水环境中新污染物的极难降解性,本研究选择全氟烷基和多氟烷基物质(PFASs)作为技术验证的对象。本研究的目的是:(1)确定不同酸碱环境下PFOA在PAA活化反应体系中的衰变和除氟能力;(2)检测并分析完整多通道ETP所涉及的传播方向和过程,以及PAA分子在PAA-AWT中除均溶过氧键外的功能;(3)确定PAA/NC系统中的关键活性物质和脱氟途径。
图文导读
PFOA的分解与脱氟效率

图1: (a) PFOA分解和除氟率的时间分布曲线(a),随pH值(b、d)和伪一阶速率常数(kobs)(c)的变化(插图:kobs时间分布曲线)与初始pH作用关系。
图1a为PAA/ND_800处理后PFOA的分解率(deC%)与除氟率(deF%),PFOA在25 min内完成衰减,deF%达到72%。在单独比较ND_800和PAA的有效性时,前者略有优势,可归因于一个经常被报道的机制,即NC颗粒表面对全氟辛烷磺酸分子的强吸附,促进C―F键的拉伸,从而有助于除氟。此外,PAA在ND_800和ND_800+PFOA体系中的分解率分别为38%和55%(图S1),结合ND_800单独对PFOA的温和降解(图1a),这一结论似乎支持了氧化剂的活化有助于碳基催化剂吸附效率的结论。为了确定PAA是否促进ND_800对PFOA的分解,进行了不同时间间隔Na2S2O3对PAA的淬火实验。如图S2所示,PAA的猝灭与PFOA降解的停止几乎是同步的,说明是ND_800激活PAA的分解而不是PAA改性对ND_800的吸附增强了PFOA的去除。然而,PAA分解过程中产生的氧化自由基是否显著影响PFOA的去除还有待进一步验证。值得注意的是,PFOA的deC%和deF%都表现出明显的pH依赖性(图1b, 1c, 1d),在酸性和中性条件下(pH≤7.5), kobs相当,范围为0.0101 min-1至0.0161 min-1。然而,一旦pH超过7.5,速率常数增加了近三倍,在pH为9.5时,效率和速率达到峰值,kobs为0.1995 min-1。还研究了PAA中存在的少量H2O2浓度的影响,发现可以忽略不计(图S3)。在强碱条件下会促进PFCs的分解,但在酸性和中性条件下的deC%和deF%的部分效果也可归因于氧化PAA/NDs AWT中产生的自由基的氧化作用。
电子转移路径

图3:线性扫描伏安法测量的电流曲线(a),开路电位测量的电子传递状态(b),计时电流法在不同反应体系中获得的EDC (c)和EAC (d)的变化和电流传递原理图。
为探究PAA/ND体系在碱性水样中的反应种类和反应机理,参考了Guo等人最近的研究,发现电子转移和自由基氧化途径可以同时发生在同一个AWT系统中,该途径的发生受催化氧化配合物与污染物之间的能隙和有机污染物的亲电指数影响。基于此,我们初步假设效率的提高是由于PAA分解过程中非自由基电子转移的贡献,类似于Kim等人提出的PAA-AWT通过电子从目标物质转移到自由基而降解芳香族化合物。PAA的活化和PFOA的降解涉及电子从PFOA(电子供体)转移到PAA(电子受体),ND作为有效的电子转移介质。ND表面的电子聚集能力已在其他碳基AWT体系中得到广泛验证,如过硫酸盐。图3a所示的I-V曲线显示了系统的预期电流产生,表明具有良好的导电性(图3b),这是降解过程中ETP的初步证据。进一步的研究表明,电子转移途径可能是PAA基氧化的主要途径。如图3b所示,在PAA注入系统后,ND_800功率涂覆电极的OCP迅速出现,表明ND_800表面与PAA之间发生了电荷转移。电流强度随着PAA剂量的增加而增加,最佳值(150µM)与PFOA降解效果一致(图1d)。ND_800表面与PAA之间的电荷转移使电位升高,其中PAA作为电子受体。然而,加入PFOA后,系统的稳定状态被破坏,导致电位显著增加+0.24 V。这一结果不同于由有机物与其周围环境之间的电位差引起的ETP非自由基途径,为目标有机物作为电子受体的功能提供了相应证据。单独添加PFOA时观察到的电位增加(+0.17V)(图3b)进一步说明了从NDs到PFOA的电流转移。这些发现表明PAA/ND_800在碱性条件下(pH>7.5)对PFOA的处理效果与酸性条件下(pH>7.5)有机电子迁移的非自由基路径和活性物质的直接攻击不同,可归因于多相系统传递的稳态电流。然而,有两个方面尚不清楚:1)PAA在电流传输中的具体作用;2)瞬时电流是否是PFOA分解的直接原因。

图4:(a) DMPO在PAA/ND_800体系中吸附5 min (CDMPO = 50 mM, pH 3.5±0.3)时自由基加合物的ESR光谱;(b)硒酸盐的添加对deF%的影响;(c) PAA和ND向不同反应底物的连续电子转移示意图。
图3c和3d显示,分别注入PAA和PFOA后系统中出现了明显的电流,尽管目前的EDCs结果并不典型,但重申了PAA/ND_800相互作用中电子的产生。在机污染物降解的ETP非自由基有过程中,有机物的引入有望促进电极上的电子积累,导致EDCs的增加,正如Wang等人进行的PAA/活性炭(PAA剂量为0.065-0.39 mM, pH = 3-9)降解SMX (0.079 mM)所观察到的结果。有趣的是在本研究中,添加PFOA (0.25 mA)后EDC的增量约为添加PAA (0.45 mA)后的一半。这表明PFOA的加入使流向电极的电流发生了转移,表明ND_800作为电子供体或介质促进了电子传播方向向PFOA方向转移。这一现象阐明了在将PFOA引入系统后观察到的OCP增加。Wu等人证明,碳质材料既可以向过氧化物提供电子以生成活性物质,也可以接受来自活性物质(如•OH和SO4•)和过氧化物的电子,从而在催化剂中形成电子通量。然而,在引入PFOA后,该体系中涉及PAA分子和ND表面的电子转移方向似乎立即发生了变化。如图3c所示,PAA的加入不会产生明显的EACs信号,但PFOA的引入显著增加了电极附近的电流,强度几乎增加了五倍。这表明PAA和ND_800之间的ETP从ND_800→PAA到PAA(作为电子供体),一旦引入能够作为电子受体的目标有机物质,将电荷转移到ND_800表面(作为电子受体)。基于这些发现,PFOA在PAA/ND_800体系中降解过程中的ETP可以描述如下(图4c):1.在PAA/ND_800 AWT过程中,PFOA作为电子受体注入后,PAA分子(或其内部的O-O键)转变为电子发射器或中间体。2. ND_800接收PAA裂解发出的电子通量,同时也作为中介将更高能量的电子流送入水中或直接射向目标。3. PFOA仅作为电子受体起作用,在PAA/ND_800处理框架内促进电子流通。
虽然EAC测量可以证明系统的还原性,但PAA/ND_800系统中导致PFOA降解的主要还原性物种仍未确定。ND作为一种半导体,利用其负电子亲和(- 0.8 ~ - 1.3 ev)和高导带(CB)位置可以在高能激发下将水合电子(eaq-)发射到周围的水相。因此,进行了EPR捕获试验以检测eaq-的存在。在含有PAA、ND_800、DMPO和PFOA的反应混合物中检测到质子化DMPO-电子(DMPO*H)自旋加合物,观察到自旋哈密顿参数为DMPO*H自旋加合物,强度比为1:1:2:1:1:1:1:1 (图4a)。不出所料,EPR信号受PFOA浓度的影响,与EDC和EAC的结果一致,PAA和ND_800之间的电子转移速率(或诱导的eaq−)受到目标(PFOA)浓度的影响,这表明在添加目标之前,PAA和ND_800之间的电流没有释放到水溶液中。然而,考虑到PFOA的电子接收能力,一部分诱导的eaq-将被释放到水溶液中,因此,EPR可能检测到发射到水溶液中的多余电子或PFOA直接接收的电子通量,需要进一步调查和分析。
活性物种鉴定
EPR光谱结果强烈表明,PFOA的加入明显阻止了氧/质子与eaq-的相互作用,与之前的结果一致,PFOA作为eaq-的靶标,也为通过ND_800表面迁移产生电子提供途径。为了进一步验证体系中eaq-的持续产生对PFOA的还原作用,考虑到CdSO4与eaq- (eq 3) 的高速的二级反应动力学(k = 6.4×1010 L mol-1s-1),选择其作为eaq-探针进行实验。尽管SO42-的标准氧化还原电位(E0 vs SHE)为0.172 V,但硫酸盐的电化学还原几乎无法实现,这表明硫酸盐在淬灭eaq-方面没有显著作用。然而,与预期相反,CdSO4的引入对PFOA分解的影响最小,为18%,对F%的抑制作用为8%(图S8),由此对eaq-的连续或瞬间形成产生怀疑。因此,我们使用N2O进行了另一个典型的eaq-探针实验,N2O与其反应具有很高的速率常数k=9.1×109L mol-1 s-1 (eq 4) 。数据表明,在N2O饱和溶液中去除PFOA没有明显的抑制作用。这些结果充分表明,PAA/ND_800系统对PFOA作用的活性降解物种不包括eaq-。

电子通过双极(偶极)反应被水捕获,并被水“笼”包围,成为可溶的电子。在形成eaq-之前的电子称为预水合电子(epre-)。一些早期的结果表明,epre-可能参与了化学还原,而不仅仅是eaq-的产生,并且一些溶质对epre-的反应速率因子比eaq-的反应速率因子高。已经证实,硒酸盐与epre-(eq 5)的反应速率系数极高,而不影响水解或氧化分解过程中其他物质的产率。Simon M. Pimblott等人详细研究了三种电子清除剂(SeO42−、NO3−和Cd2+)的水辐射化学。Cd2+是一种具有快速(eaq−+ Cd2+)反应的高效eaq−清除剂,实验结果不明显;NO3−是一种效率较低的eaq−清除剂,在浓溶液中对eaq−的清除效果明显;而对于低效的eaq−清除剂SeO42-,即使在稀溶液中也有明显的eaq-预清除作用。结合PAA/ND_800对PFOA降解的还原性能和eaq-的检测结果,选择硒酸盐作为捕集剂,探讨epre-在PAA/ND_800降解PFOA中的作用。图4b所示的结果为我们对epre -的预测提供了证据。硒酸盐离子(CSe)浓度直接影响PAA/ND_800体系中PFOA的deF%和分解,低硒酸盐浓度(1 mM)可使deF%从95%降低到9%。幸运的是,CSe与deF%呈现出令人难以置信的关系。当CSe浓度大于0.75 mM时,添加量与deF%呈良好的线性关系(R2 = 0.98);然而,从0.75 mM开始逐渐减小后,尽管与deF%呈正相关,但出现了线性错配(R2 = 0.79)(图S9a和S9b)。这表明硒酸盐消除的活性物质并不完全是导致deF%的主要活性物质。考虑这一现象的原因是高剂量的硒酸盐在去除epre-的同时也能清除eaq-,而这两种电子的反应速率却大不相同(公式5,6)。考虑到CdSO4和N2O两种eaq-探针对工艺性能的影响差异以及N2O对系统中eaq-信号的抑制作用(图S10),可以判断,一旦CSe浓度超过0.75 mM,经PFOA处理的PAA/ND_800工艺中的epre-被彻底去除,eaq-被同时捕获。最重要的是,当PAA分解形成电子通量并转移到ND_800表面时,它立即与吸附在ND_800表面的PFOA分子发生反应,形成可溶的eaq-;因此,在PAA/ND_800体系中,epre-是PFOA deF%的主要活性电子,其他电子向水中释放形成溶解的eaq-, eaq-对PFOA deF%也有一定的促进作用,可认为是次级活性电子。
小结
eaq-对PFOA以外的全氟化合物和氟化端粒也表现出高反应性。然而,现有的依赖亚硫酸盐、碘化物等的高级还原体系(ARPs),由于惰性盐残留物的积累,通常伴随着出水中总溶解固体(TDS)的增加。PAA在1~ 15mg /L的剂量范围内已被一些水厂成功应用于水和/或废水消毒,而不影响后续处理效果或增加额外的控制环节。本研究提出的基于PAA的ND-AWT体系消除了在建立AWT过程中对水的不利影响,揭示了PAA-AWT连续电子转移的非自由基机制,不同于以往报道的单重态氧、单电子或双电子单路转移机制,并确定了epre-在eaq-溶解之前的重要作用。为了推进PAA基和/或预氧化- AWT的实际应用,进一步的研究工作应考虑:1.阐明真实水基质的背景因素对还原效果的影响,如溶解氧、NO3-等与eaq-的高反应速率指标;2. 新污染物还原产物(包括短链PFCs)的环境影响及后续处理。例如,PAA/ND_800产生的还原性TPs,如果在其上保留了丰富的还原官能团,则可以通过某些化学氧化过程有效地降解,或者当ARPs提高了这些化合物的生物降解性时,则可通过生物降解进一步去除。总之,PAA诱导AWT的非电子转移还原机制为去除水中脂肪族类新污染物提供了思路。
本项目得到了国家重点研发计划青年项目“水中新污染物等风险物质高通量识别与传感前沿技术研究”的资助。
作者简介
第一作者

薛雅内: 清华大学环境学院助理研究员;任中国环保机械行业协会紫外专委会委员,纳滤膜产业联盟副秘书长等职务。主要从事水中新污染物去除与传感、紫外线设备研发应用、纳滤膜技术。获2024年日内瓦发明金奖、2023年中国环保机械行业协会技术创新一等奖(排名第二)、2023年清华大学博士后支持计划、2018年黑龙江省科技创新奖(排名第二)、哈尔滨工业大学“HIT TIMES”优秀论文等奖励;主持/参与国家重点研发计划、国家重点研发计划青年科学家、国家重点研发计划国际合作等项目;发表SCI收录论文15余篇,参编专著3部,团体标准2项。
通讯作者

孙文俊: 清华大学环境学院副研究员,清华苏州环境创新研究院现代净水团队PI,加拿大西安大略大学兼职教授;任中国环保机械协会紫外线专委会主任委员兼秘书长,中国生产力协会水与健康委员会副主任委员,IUVA董事会成员等职务。获2024年日内瓦发明金奖、2023年国际紫外线协会工最佳紫外线工程奖、2023年中国环保机械行业协会科技进步一等奖、2021年中国环境保护产业协会环境科技进步一等奖、2020年北京市科技进步二等奖等奖项(均排名第一);在UV技术研究与应用、给水深度处理、污水高效处理、节水技术等领域取得多项创新成果,主持/参与国家级科研项目10项,出版专著5部,编制国家标准4项,发表论文200余篇。