罗胜联教授团队:硫离子掺杂促进WO₃纳米线高效光催化降解甲苯
摘要
通过外加H2O2溶解固态钨网上表层钨单质并产生过氧钨酸,同时在前驱体溶液中加入硫代乙酰胺,结合水热合成法与煅烧法制备得到S2-掺杂的WO3光催化纳米线(标记为S-WO3),构建了氧空位介导的高活性亲水/亲氧光催化反应系统,深入探讨了S掺杂及氧空位对促进原始WO3光催化性能及自由基生产能力的内在机制,并通过一系列甲苯的光催化降解实验验证了其污染治理能效。结果表明:S-WO3在90min内对甲苯的去除率为91.7%,是原始WO3的3.3倍;其对应的CO2转化率为90.1%,是原始WO3的15.8倍。X射线衍射(XRD,X-ray diffraction)和电子扫描电镜(SEM,scanning electron microscope)结果显示,S2-掺杂会引起WO3晶格畸变和应力场导致晶体重新定向生长,使WO3从纳米块状结构转变成纳米线状结构,因此S-WO3纳米线具有比原始WO3更大的比表面积,光吸收能力增强。此外,电子顺磁共振(EPR,electron paramagnetic resonance)结果表明:S2-掺杂也会引起WO3晶格中氧空位产生,并作为电子陷阱捕获光生电子,促进载流子分离,提高光电转换效率。同时,与原始WO3相比,S-WO3亲水性变大,能更加有效地吸附水分子和氧分子,增大被空穴氧化和光电子还原概率,生成强氧化性羟基自由基(·OH)和超氧自由基(·O2-)。综上,S2-掺杂显著增强了WO3降解矿化甲苯能力。且由于S-WO3直接生长在钨网上,这种自支撑特性使其物化性能稳定,连续5次循环实验,其甲苯降解效率未见衰减,且反应过程中不产生苯等有毒中间体。相关研究结果展示了S-WO3催化剂作为高效持久降解VOCs的催化能力和实际应用前景。
研究背景
甲苯是一种广泛使用的有机溶剂,在化工合成、涂料、树脂、染料、炸药和农药等领域有着广泛应用。甲苯的主要污染源包括化工产品、与汽油有关的排放、食品行业的污染等。在建筑材料的有机溶剂中大量存在,如油漆、防水材料等,同时在日常生活中也用作装饰材料、人造板家具、黏合剂、空气消毒剂和杀虫剂的溶剂。甲苯对人体健康具有较大毒性,对皮肤和黏膜有刺激作用,对中枢神经系统的作用比苯强。长期接触甲苯可能导致神经衰弱综合征、生殖系统问题、甚至膀胱癌。此外,甲苯对空气、水环境及水源可造成污染,对鱼类和哺乳动物构成威胁,甲苯的挥发性使其在环境中通过空气和水体之间的再循环而广泛分布。在传统的甲苯治理方法中,物理吸附和化学氧化等技术因其能耗高、易产生二次污染等问题而受到限制。因此,开发绿色环保的甲苯净化技术是当前环境领域的研发热点。光催化技术因其能够在室温下通过可见光激发产生氧化性自由基,将有机污染物矿化成无害的小分子,且不消耗化学试剂,被视为一种环境友好型的甲苯污染绿色治理手段。
WO3作为一种窄带隙(2.5~2.7 eV)光催化剂,可以吸收太阳光谱中波长在500 nm以下的可见光,产生的光生电子-空穴对具有强的氧化、还原能力,对推动后续一系列的化学反应具有积极作用。但是,单相WO3中,光生电子-空穴对复合率高限制了其在光催化领域的深度应用。此外,光催化激发自由基生产需要通过O2/H2O和电子/空穴间的氧化还原反应来实现,从这个角度来看,如何提高WO3的光催化效率并促进O2/H2O在光催化剂表面吸附是促进WO3高效降解甲苯的关键。氧空位可以调节和重建WO3的电子结构,增强电子在光催化剂表面转移,并作为电子收集中心来促进O2/H2O在光催化剂表面吸附。因此,研究人员一直在努力通过在WO3上构建缺陷工程或掺杂/异质连接来增加氧空位暴露密度。
大量研究表明:通过金属和非金属掺杂可以在WO3晶格中引入额外的缺陷态,这些缺陷态在能带中形成浅能级,有效捕捉光生电子或空穴,减缓其复合速度来提升其光催化性能。光生电子-空穴对的复合与转移是决定催化剂活性的重要影响因素。一般情况下,光生电子-空穴对的复合速率远高于其在界面传输时的速率,通过缩短迁移路径减少界面的迁移时间,能够有效提高载流子的分离效率。例如,有研究通过Cu元素的原子掺杂,实现了WO3的上转换与电荷分离性能的协同提高,显著增强了其在红外光下催化四环素降解的能力。但由于过渡金属的原子半径差距及外层电子数量较多,通常导致对WO3的掺杂成功率较低,限制了催化活性位点数量和催化性能。但与此同时,贵金属掺杂(如Pt、Au)昂贵的成本和稀有性限制了实际大规模应用的可行性。而相比较于金属掺杂,非金属掺杂能够更直接有效地改变WO3的带隙能量,形成新的能级结构,提升其可见光利用率。在众多非贵金属元素中,相较于N和C等元素,S元素的电负性和半径适中,可以较好地与WO3形成掺杂结构并调控载流子的迁移和分离行为,从而显著延长光生载流子的寿命,提升电荷分离效率。同时,S2-掺杂可以有效增加WO3中氧空位的含量,氧空位可以通过吸引电子,抑制电子-空穴对的复合,提升光催化氧化降解空气中污染物的效果。另外,S2-掺杂产生的氧空位还有助于增强水分子与氧气分子在S-WO3表面上的吸附,产生更多活性中间体(如·OH与·O2-等)。
基于以上背景,在该工作中,首先用双氧水浸泡钨网,强氧化性双氧水可将单质钨网的表层氧化溶解为过氧钨酸(H2WO4),成为WO3的前驱体;然后外加硫代乙酰胺为S2-的前驱体。相关溶液与钨网经水热反应后,将生长有硫掺杂WO3(S-WO3)的钨网在氩气环境中600 ℃煅烧以提高其结晶度,从而制备初S-WO3催化网并应用于甲苯光催化降解。通过对比研究,探索S2-不同掺杂量对WO3降解甲苯效率的影响规律。此外,以S-WO3为光催化剂探究了其对甲苯和甲醛混合VOC气体的处理能力。通过改变条件(甲苯初始浓度、循环次数、湿度)测定了其对甲苯的降解性能,并基于电子顺磁共振、接触角、BET和紫外可见漫反射等表征结果,深入探究了S-WO3的光催化反应机理。
01 实验部分
1. 化学试剂与材料
过氧化氢(H2O2,30%,AR)、硫代乙酰胺(C2H5NS,AR)、丙酮(CH3COCH3,AR)购于西陇科学公司。无水乙醇(C2H5OH,AR)购于上海振兴化工有限公司。钨网(纯度99.8%,100 目)由晨屹丝网有限公司提供。所有试剂均溶解在去离子水中。
2. WO3与S-WO3的制备
WO3的制备:将经预处理(在乙醇溶液中超声振荡15 min去除表面油脂)的钨网片浸入60 mL去离子水中,随后加入160 μL的H2O2,超声振荡10 min。超声结束后,将得到的液体和钨网片一起转移至100 mL的反应釜内,通过电热鼓风干燥箱将反应釜加热到160 ℃,并且在该温度下保持12 h。自然冷却后,取出钨网片用去离子水对其进行冲洗。洗净后的钨网片在经过干燥后,将其放入管式炉中,在氩气的氛围下,600 ℃下煅烧90 min,自然冷却得到WO3催化剂。
S-WO3的制备:将预处理过的钨网片浸入60 mL去离子水中,随后向其中加入160 μL的H2O2,超声振荡10 min。接着,称取一定量的C2H5NS (30, 60, 90, 120 mg)加入到上述前驱体溶液中,超声振荡5 min。超声结束后,将得到的液体和钨网片一起转移至100 mL的反应釜内,通过电热鼓风干燥箱将反应釜加热到160 ℃,并且在该温度下保持12 h。自然冷却后,取出钨网片用大量的去离子水对其进行冲洗。洗净后的钨网片在经过干燥后,放入管式炉中,在氩气环境下600 ℃煅烧90 min,自然冷却得到SWO3催化剂。
3. 表 征
为了测定催化剂的颗粒形貌、孔隙结构、表面粗糙度等形貌特征,采用SEM对催化剂进行表征;通过透射电子显微镜(TEM,transmission electron microscopy)观察WO3和S-WO3催化剂的晶格结构与畸变、缺陷;使用XRD技术分析催化剂的晶相结构;通过X射线光电子能谱(XPS,X-ray photoelectron spectroscopy)分析催化剂材料表面元素的化学态和价态;利用紫外可见漫反射(UV-vis DRS,ultraviolet-visible diffuse reflectance spectroscopy)测试WO3和S-WO3光催化剂的光吸收范围和带隙宽度。利用紫外光电子能谱(UPS,ultraviolet photoelectron spectroscopy)探测WO3和S-WO3的价带结构;利用EPR技术检测WO3和SWO3光催化剂缺陷中未成对的电子,同时EPR还可以探测在催化反应过程中产生的自由基;采用接触角仪评估WO3和S-WO3光催化剂表面的亲水性;利用气相色谱测试光催化过程中甲苯浓度变化以及其转化成CO2的程度;应用电化学工作站测定WO3和SWO3光催化剂的阻抗和光电流强度;通过瞬态荧光光谱仪(TRPL,time-resolved photoluminescence)检测WO3和S-WO3的荧光寿命,利用稳态荧光光谱(PL,photoluminescence spectroscopy)检测WO3和S-WO3分离电子-空穴对的能力。
4. WO3及S-WO3光催化降解甲苯性能测试
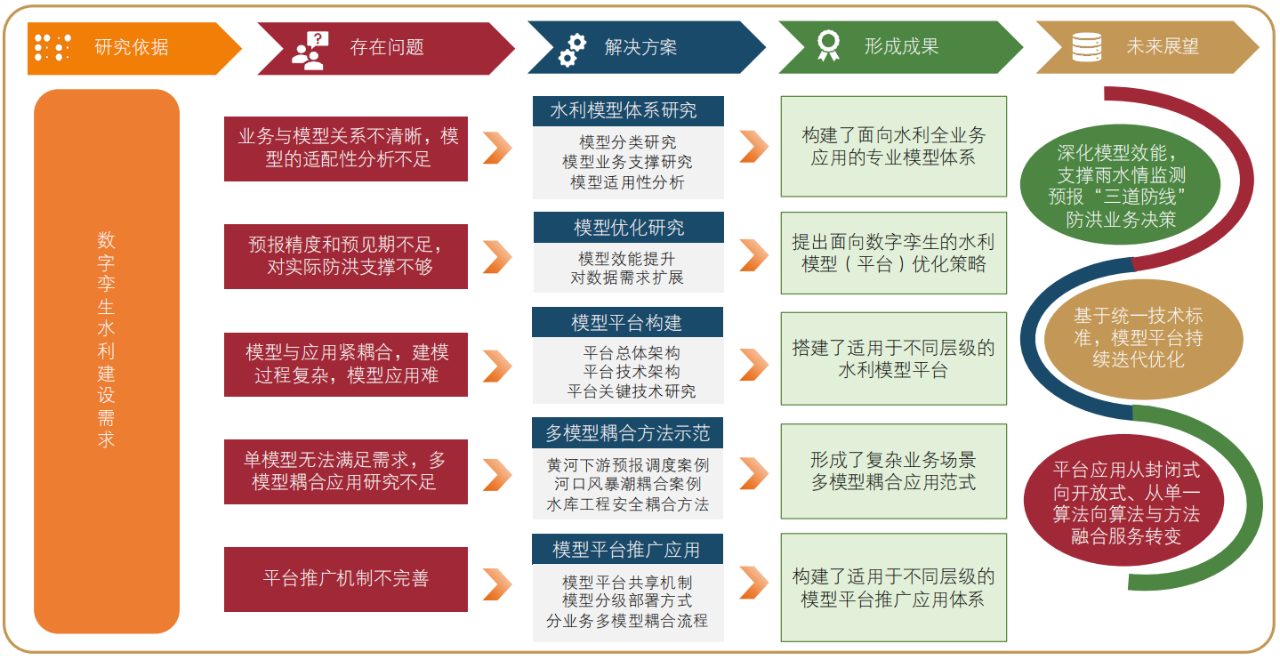
光催化甲苯在体积为450 mL石英反应器(图1)中进行(光源为氙灯,模拟太阳光源),由石英窗、单斜孔、进样口以及冷凝水进出口组建而成,确保在实验过程中可以有效控制反应条件。实验中氙灯放置在石英窗上方约25 cm处。单斜孔能够调节反应器中气体的注入与排出。进口1为进样口与取样口,也能放置感应探头。在开始实验之前,通过放置干燥剂与进口1的感应探头配合,检测并适当调控反应器内湿度,防止过多水分对反应的干扰。催化剂的面积为5 cm×15 cm,通过折叠放置在适当的位置,确保光照均匀覆盖在催化剂表面。在打开氙灯之前,用量程为1 μL的玻璃取样器抽取0.3 μL液态甲苯注入石英反应器中,用吹风机对反应器进行加热,使甲苯快速挥发,最终气态甲苯浓度为30×10-6。光催化反应开始之后,为了实时了解甲苯的降解情况和CO2的生成情况,应用气相色谱仪对反应器内的甲苯浓度和CO2浓度进行测定。

图1 光催化降解甲苯的反应器示意
02 结果与讨论
1. 硫离子掺杂对WO3晶相、形貌、结构的影响
图2a、2b分别为纯WO3催化剂和硫代乙酰胺用量为90 mg条件下制备的S-WO3催化剂微观形貌。可知:经过硫掺杂之后,WO3的形貌渐渐从不规则的纳米块状结构演变成纳米线状结构,WO3纳米线长1~2 μm,直径约100 nm。与WO3纳米颗粒相比,纳米线状结构具有更大的比表面积和更高的活性位点密度,有利于光催化界面对目标污染物质、水和O2的吸附,促进随后的光催化反应并提高甲苯降解效率。

图2 WO3和S-WO3光催化剂的形貌结构与元素分布分析
图2c,2d分别为WO3和S-WO3(020)晶面的晶格条纹。值得注意的是,WO3的(200)面和S-WO3的(020)面的晶格条纹间距分别为0.365, 0.369 nm。这说明硫元素的引入导致了WO3(200)晶面间距增加,这一现象与XRD光谱中2θ角度的低角度偏移是一致的。图2d中用黄色的圈标出的一部分图像进一步证实了这一点。在这些图像中,可以观察到由于硫的引入,WO3发生晶格膨胀、畸变、缺失,同时引入一些缺陷。此外,通过TEM图像(图2e)的元素分布,可以观察到钨(W)、氧(O)、硫(S)等元素在纳米线上的均匀分布情况。这种分布模式佐证了S在WO3中的成功掺杂。
图3为未修饰WO3和S-WO3光催化剂的XRD图。S-WO3的XRD图谱中,2θ为40.3°、58.3°和73.2°处观测到的衍射信号峰,分别隶属于W基底的(110)、(200)和(211)晶面,与W(PDF#04-0806)的标准卡片相吻合。其中,2θ为23.1°、23.6°、24.4°和34.2°处观测到的衍射信号峰分别与WO3的(002)、(020)、(200)和(202)4个晶面对应,这与正交WO3结构(PDF#43-1035)的标准卡片相吻合。伴随着S2-的引入,4个特征衍射信号峰都发生了一定程度的变化(下降或变宽),这意味着S2-的掺杂降低了WO3的结晶度。通过对比2θ为32.5~35.5°范围内的(202)衍射信号峰(图3b),发现硫掺杂之后,峰位置发生了向左偏移,即2θ的值变小了。通过将θ值带入布拉格定律d(hkl)=λ/(2sinθ)中,可以计算出WO3晶面间距d(202)增加。因此可以合理推断,硫的引入导致了WO3的晶格膨胀。

图3 WO3和S-WO3光催化剂的晶相分析
2. WO3及S-WO3催化剂XPS能谱图分析
对WO3和S-WO3催化剂进行XPS表征,结果如图4a所示。可知WO3和S-WO3催化剂中均含有W、O元素。S-WO3中S元素含量较低,导致其光谱信号较弱,仅在165 eV处有个小峰,对应于硫元素的2p轨道能级。O1s的高分辨XPS光谱图如图4b所示。初始的WO3在529.41 eV处的特征峰代表WO3中晶格氧;而在S-WO3的高分辨O 1s光谱图中有3个特征峰529.43, 531.21, 534.08 eV,分别归属于WO3中的晶格氧、吸附氧、羟基氧,其含量占比分别为49.43%、34.18%、16.39%。与原始WO3相比,SWO3中羟基氧与吸附氧的出现说明了S掺杂导致WO3的晶格氧部分缺失,形成氧缺陷并促进O2和H2O在S-WO3表面吸附,这为活性自由基的产生提供了基本基础。图4c中W 4f的高分辨XPS光谱显示初始的WO3在35.81, 37.62 eV的结合能分别对应W 4f7/2和W 4f5/2轨道的光谱支项;而S-WO3的W 4f7/2和W 4f5/2所对应的结合能分别为35.57, 37.38eV,同时在33.44, 36.01 eV处出现了+5价钨的特征峰。以上结果显示,W的4f轨道对应电子结合能降低。这种负偏移通常表明W原子价态的降低,即从+6价态向更低的价态转变,这是由于S掺杂引入的额外电子可以填充部分钨原子的4f能级,导致该轨道上电子结合能下降。S 2p的高分辨XPS光谱如图4d所示,原始WO3的未观测到硫的特征峰;SWO3的S 2p图谱中161.43, 162.64 eV处出现的特征峰被归属于硫化钨;而在166.73, 167.86 eV则归属于前驱体中硫代乙酰胺中的S,这佐证了上述TEM和XRD的分析结果。

图4 WO3和S-WO3光催化剂的X 射线光电子能谱表征
3. WO3与S-WO3光催化剂的光电性能表征
在光催化氧化的过程中,光生电子(e-)和光生空穴(h+)必须有效分离并迁移到达催化剂的表面,才能在催化剂表面上参与后续的氧化还原反应,从而实现甲苯的降解。光电流强度是衡量催化剂光生载流子分离效率的直接指标,电化学阻抗则可以提供关于催化剂导电性和载流子迁移速率的信息。测定由WO3、S-WO3制成的电极上的光生电流密度,相关结果如图5a所示。可知WO3催化剂的光电流响应约为0.3 μA/cm2,S-WO3的光电流响应约为0.6 μA/cm2,这表明S2-的引入强化了WO3的光生载流子的分离效率。此外,在多次连续光照-黑暗循环过程中,SWO3光电转换性能稳定。电化学阻抗图能表征光生电子在界面传导所遭遇的电阻,即界面导电性由阻抗弧的半径衡量:半径越小,内阻越小,光生电子在光催化界面的转移速率就越快。经过硫掺杂的WO3阻抗的半径明显变小(图5b),说明S2-能在WO3中形成掺杂能级,减小了光生电子在WO3中的迁移电阻,使其尽快转移到催化界面,从而降低了与光生空穴在催化剂内部的复合概率,提高了光电转换效率。

图5 WO3和S-WO3光催化剂的光电化学性能及比表面积、孔径分析
为了深入理解WO3和S-WO3催化剂在光催化过程中光生载流子的动态行为,采用稳态荧光光谱和时间分辨荧光衰减光谱技术对其进行性了详细分析,如图5c所示。在稳态荧光测试中,可以观察到初始的WO3在470 nm处展现出较高的荧光强度,这表明其光生载流子复合较为显著。而当S2-掺杂入WO3之后,稳态荧光强度显著变弱,这一变化反映了S2-掺杂能够增强光生载流子的分离,降低光生电子-空穴对复合的概率。稳态荧光强度降低的趋势与之前光电流响应和阻抗的测试结果一致,相关结果都证明了S-WO3催化剂在光催化过程中载流子分离效率的显著提升。图5d为WO3及S-WO3催化剂的时间分辨荧光衰减光谱测试,WO3和S-WO3的平均荧光寿命分别是0.32, 0.78 ns。长寿命的光生载流子对于光催化过程起着重要作用,因为这些载流子可以迁移到WO3表面,与吸附在其上的水和O2相互作用,产生活性氧物种以氧化污染物。
对于光催化降解挥发性有机物而言,催化剂的比表面积越大,提供的催化活性位点就越多,催化效率越高。介孔孔隙体积同样影响着催化剂的催化性能,不仅关系着污染物的吸附能力,也影响污染物在催化剂表面的扩散速率。较大的孔容度可以促进污染物在催化剂内部传输,从而加快反应速率。因此,本工作还研究了WO3和S-WO3的N2吸附-解吸等温线和孔径分布,如图5e、f所示。可知:WO3和S-WO3催化剂样品都具有IV等温线与H3型滞后环,这意味着2种催化剂都是介孔结构。比表面积、孔径和孔体积的值总结见表1,相比于初始的WO3,S-WO3的比表面积和孔体积均大幅增加。较大的比表面积有利于WO3催化剂上活性位点的充分利用,较大的孔体积增强了水分子、O2、污染物的吸附和运输。
表1 WO3与S-WO3的孔容积、孔直径和比表面积

4. 不同S-WO3光催化剂高效降解甲苯与甲醛
氙灯照射下,探究了S2-掺杂量对WO3催化剂光催化氧化去除甲苯性能的影响,如图6a、6b所示。可知:未改性的WO3在90min内对甲苯的去除率为28%,相对应的CO2转化率为5.7%。相比之下,随着硫含量在WO3中的不断增加,S-WO3的甲苯降解率随之加快,CO2产量逐渐增高。当硫代乙酰胺的添加量达到90 mg时,S-WO3对甲苯的降解率和CO2转化率达到最高,此时90 min甲苯的降解率为91.7%,是纯WO3的3.3倍;相对应的CO2转化率达到了90.1%,是未改性WO3的15.8倍。继续增加前驱体中硫代乙酰胺的添加量到120 mg,可观察到S-WO3对甲苯的光催化效率开始下降。这可能是由于过多的S2-影响光催化剂的晶体结构和表面形貌,导致催化活性降低。根据这些结论,确定S2-的最佳掺杂量为90 mg。图6c为最佳S-WO3光催化剂降解不同初始浓度(12×10-6、30×10-6、51×10-6、69×10-6)甲苯的去除效率。可知,随着甲苯初始浓度的不断升高,对应的甲苯降解率有微弱下降,这可能是由于甲苯矿化导致的催化剂表面积碳所致。实际生活工作场景中,甲醛和甲苯是室内VOCs污染的主要成分,两者共存是常见现象。因此,同时去除2种气体混合污染物的光催化剂能更好地应用于实际生活当中。基于此,使用初始WO3和S-WO3催化剂对甲苯和甲醛的混合物进行了光催化降解,评估结果如图6d所示。在90 min内S-WO3光催化剂对甲苯和甲醛的降解率分别为72%和79%。而WO3在90 min内对甲苯和甲醛的降解率仅为22%和28%,这一差异也表明S2-掺杂对WO3光催化剂性能的提升作用显著。

图6 不同S-WO3光催化剂降解甲苯、甲醛的性能测试
在光催化氧化甲苯的过程中,水分子是强氧化性·OH前驱体,光催化反应器中的相对湿度与甲苯的降解率密切相关。图6e为S-WO3在不同湿度环境下对甲苯降解性能,当光催化反应器中的相对湿度(RH)从20%逐步上升至60%时,甲苯的降解率呈现出与湿度相呼应的递增趋势,由42.65%上升至91.7%,这是因为吸附在光催化界面上的水分子浓度增加,有更多的·OH在S-WO3光催化表面上生成并参与甲苯分子的氧化分解,导致甲苯开环速率变快,去除更迅速。而相对湿度从60%上升至100%时,甲苯的降解率反而出现了下降,由91.7%下降至51.23%,可能原因是过多的水分子在催化剂的表面形成了水膜,与甲苯分子形成竞争吸附,阻碍了甲苯分子跟S-WO3催化剂的界面接触,降低了甲苯分子与·OH的反应概率,导致S-WO3催化性能下降。此外,S-WO3催化剂的稳定性是其成为空气净化领域中具有应用前景的关键基础。S-WO3催化剂在经过5次循环实验后,对甲苯降解率从92%略微下降至90%(图6f),这一小幅度变化反映了S-WO3光催化剂能够在长期运行过程中保持较高的活性和稳定性。总体而言,S掺杂WO3能够有效促进水分子吸附及自由基生产,对甲苯具有优异的降解矿化性能。
5. 光催化机理研究
为了探究S2-掺杂WO3在降解甲苯中的作用机制,采用了低温EPR技术来测定氧空位(OVs)、·OH、·O2-在WO3和S-WO3上的生成量,如图7a所示。初始WO3和S-WO3在g=2.003处的EPR信号归属于氧空位,其中S-WO3产生的EPR信号明显高于初始WO3,这表明掺杂样品的OVs含量较纯WO3显著增多。氧空位可以提供更多的活性位点,捕获电子,抑制电子和空穴符合,促进光生载流子的转移同时促进O2、H2O和甲苯分子在S-WO3表面吸附,构建亲水/亲氧界面,因此,氧空位的增多对于光催化性能至关重要。在图7b和7c中,峰面积比例为1∶2∶2∶1与1∶1∶1∶1的特征分别指示·OH和·O2-的EPR信号。S-WO3样品中的DMPO-·OH和DMPO-·O2-信号强度均高于初始的WO3样品,这表明S2-掺杂提高了WO3光催化剂在降解甲苯过程中活性物种的生成量。·OH和·O2-在光催化降解有机污染物的过程中起着关键作用。其中,·OH是一种强氧化剂,氧化还原电位高达2.8 VRHE,可以通过夺氢加成等方式快速进攻甲苯的甲基和苯环分子,将其矿化成水和CO2。·O2-虽然不如·OH活泼,但其较长的寿命使其能够扩散到较远的距离,可以协同·OH共同进攻甲苯分子的苯环结构。这一系列结果表明,S掺杂有效增强了WO3产生·OH和·O2-的能力,直接加强了甲苯的去除效果。

图7 WO3和S-WO3光催化剂的EPR 信号值
接触角作为一项关键的物理参数,能够揭示水分子在催化剂表面的润湿行为,进而反映催化剂的亲水性质。在光催化过程中,·OH的生成主要源于催化剂表面吸附水分子的空穴氧化过程。因此,通过精确测量催化剂表面的接触角,可以有效评估其对水分子的吸附能力,从而预测催化剂产生·OH的效率。未经改性的WO3催化剂呈现出较大的静态接触角(30.01°)(图8a),表明其相对疏水的表面特性。相对而言,经过硫掺杂处理的WO3催化剂的接触角显著降低至13.57°,这一变化清晰地指示了掺杂引入的S2-增强了WO3表面的极性,从而促进了水分子的有效吸附。增强的水分子吸附作用为·OH的生成创造了更加有利的环境。基于上述分析,可合理推测,经过硫掺杂改性的S-WO3催化剂在光催化降解甲苯等有机污染物方面的性能将得到显著提升。

图8 WO3及S-WO3光催化剂的接触角
光催化剂需要通过吸收光子的能量来激发电子从价带跃迁到导带,生成电子-空穴对,这些具有催化活性的光生载流子会参与氧化还原反应过程,推动光催化氧化反应的进行。因此,光催化剂对太阳光的光吸收特性直接影响到光催化剂是否能够充分的利用太阳能。原始WO3催化剂的吸收边为476 nm,而S-WO3催化剂的吸收边为503 nm(图9a),说明S2-的引入拓宽了WO3对太阳光的吸收范围。为了量化原始WO3催化剂和S-WO3催化剂的光学性质,通过公式Kubelka-Munk函数换算得到WO3和S-WO3的光学带隙图(图9a)。带隙(Eg)宽度是指催化剂材料中导带与价带之间的能量差,决定了催化剂能吸收光子的最低能量。原始WO3的带隙宽度为2.69 eV,SWO3的带隙宽度为2.57 eV(图9b),较窄的带隙宽度意味着能够吸收更多低能量的光子(可见光范围内的光子)。因此,较宽的吸收范围和较小带隙宽度使S-WO3催化剂在可见光驱动的光催化反应中更具优势。为了明确样品价带(VB)和导带(CB)的相对位置,采用UPS技术对初始WO3及S-WO3的VB进行测定。初始WO3的VB电位为2.89 eV(图9c),SWO3的价带电位为2.73 eV(图9d),即WO3中掺杂入S2-后,其价带电势降低。这可能是因为S2-引入取代了O2-的位置,导致WO3晶格发生局部应变和缺陷,从而减小WO3的禁带宽度。此外,S2-掺杂还可能引起WO3的电子状态和能级分布发生变化,进一步影响其禁带宽度。

图9 WO3和S-WO3光催化剂的光响应范围及带隙分析
结合图9,得到WO3和S-WO3的带隙分别为2.69 eV和2.57 eV,通过式(1)计算得到WO3和SWO3的对应导带(CB)电位分别为0.2 eV和0.16 eV,对应的光催化机理图如图10所示。导带的位置反映光生电子的还原能力,导带电势越小越有利将吸附在催化剂表面的O2还原成·O2-,这与上述结果相互印证。这些结果都证明了S-WO3能够产生足够数量的·O2-促进甲苯降解。

ECB=EVB-Eg (1)
图10 WO3与S-WO3光催化剂的机理
03 结 论
通过水热法和二次煅烧法在钨网上制备得到了S2-掺杂的WO3纳米线光催化剂(S-WO3)。S2-的引入促使WO3由纳米块状转变成纳米线状结构,增加其比表面积和光吸收位点;同时S2-也引起了氧空位的增加,氧空位能够有效捕获电子,协同形貌转变和缺陷策略可控促进了光生载流子分离,延长电子寿命,产生大量空穴,显著提升自由基生产。最优的SWO3光催化剂在90 min内对甲苯的降解率高达91.7%,矿化率同步高达90.1%,并且在进行5次循环实验后依然能够保持较好的稳定性,显示S-WO3在光催化空气污染控制技术领域有较好的应用前景。