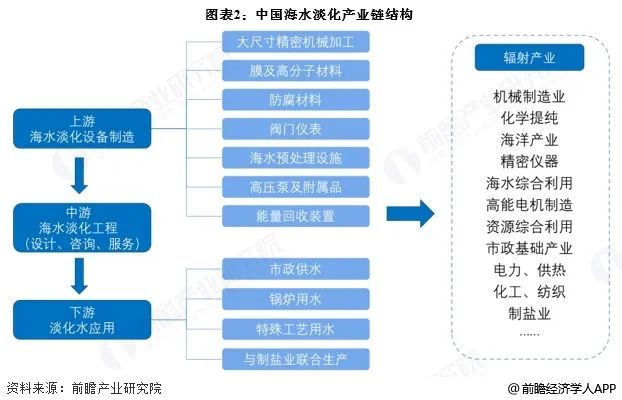
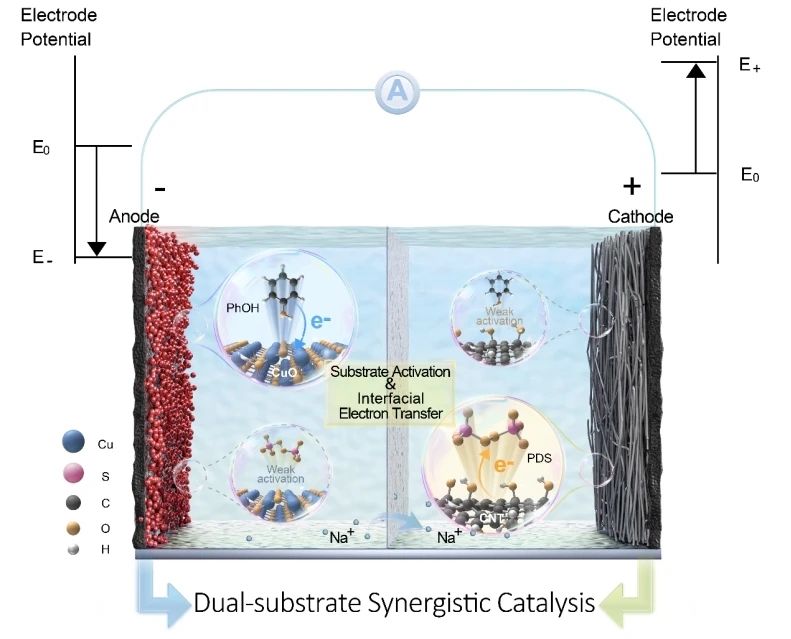
挑战传统认知:水处理高级氧化过程中 有机物是矿化还是聚合?
在水净化领域,污染物去除途径是催化剂设计、活性位点识别、构效关系建立、反应机理阐明、性能优化等的基础。近一个世纪以来,人们普遍认为,在催化氧化反应中,特别是Fenton和类Fenton反应中,有机污染物是通过降解和矿化反应途径去除的。在这些途径中,催化剂激活氧化剂生成高活性氧物种,将有机污染物分解并矿化为CO2和H2O,这一过程称为高级氧化过程(AOP)。

然而,最近对非均相催化氧化体系的研究挑战了这一传统观点,揭示了有机污染物可以通过催化剂表面的氧化偶联和聚合途径(从水相转移到固相)从水中去除,这一过程被称为直接氧化转移过程(DOTP)。

在H2O2和过硫酸盐的多相催化氧化体系中,FeOCl和Co3O4等固体催化剂通常被认为是典型的类Fenton催化剂。这些催化剂反映了均相Fe2+和Co2+的类似催化功能,能够活化H2O2或过硫酸盐生成活性羟基或硫酸盐自由基,从而降解和矿化有机污染物。然而,我们的研究表明,在这些催化氧化体系中去除有机污染物的主要反应途径不是降解和矿化。以酚类和胺类污染物的氧化去除为例,反应过程主要受偶联和聚合途径控制。此外,这种基于耦合和聚合途径而非降解和矿化途径的污染物氧化去除过程也普遍存在于具有各种氧化剂[H2O2、过氧单硫酸盐(PMS)、过氧二硫酸盐(PDS)、高价金属(例如MnOx)和非金属(例如IO4–)]、催化剂[金属氧化物、碳材料、单原子催化剂(SAC)、复合材料等]和反应条件(组分浓度、温度、pH等)的多相催化氧化系统中。
对于母体污染物,其范围甚至从微克/升范围内低浓度的微污染物到微克/升范围内高浓度的污染物。总的来说,在各种非均相催化氧化系统中普遍存在偶联和聚合途径,表明它们在污染物去除中的重要性。

热力学上,非均相催化氧化体系中偶合和聚合途径的普遍存在是合理的。矿化途径包括逐渐破坏污染物分子结构中的各种化学键,如C–C键,需要高能活性氧物种或侵蚀性反应条件。相反,聚合途径的作用相反,在C–H键氧化后,促进C–C、C–O或C–N键的形成,对氧化能力(电势)的需求较弱。最近的研究表明,在使用过氧化氢、过硫酸盐和类似试剂的多相催化体系中,羟基和硫酸盐自由基等高氧化性物种的生成具有挑战性,导致污染物氧化过程中出现许多较弱的氧化和选择性非自由基机制。因此,在非均相催化氧化体系的弱氧化环境中,一旦满足反应条件,聚合途径的发生应比矿化途径的发生优先。
总的来说,“聚合”在非均相催化氧化体系中被严重低估,因为它实际上在有机污染物去除中起着重要作用。它的重要性可以与广泛认可的水净化“矿化”途径相媲美。因此,对于催化氧化体系,应仔细研究其反应途径(聚合或矿化)。
【误解起源】
在基于H2O2、过硫酸盐、高价金属和非金属的非均相催化氧化体系中,以往对降解和矿化途径的理解通常来源于相应的均相催化氧化体系,其研究视角和方法一直被模仿。然而,非均相催化剂表面具有均相离子催化剂所缺乏的活化、稳定和积累功能,导致非均相和均相催化氧化过程存在显著差异。从均相体系到非均相体系的研究视角和方法的直接模仿可能是造成对非均相催化氧化途径误解的原因之一。另外,由于研究有机污染物反应的水相体系是稀溶液体系,污染物浓度一般在百万分之几左右,因此对产物和途径的定性定量分析以及碳平衡计算都比较困难。这可能是另一个原因的偏差,在路径确定。因此,近一个世纪以来,表面偶联和聚合途径在多相催化体系中的重要性一直没有得到认识。
【污染物去除途径识别与新技术开发】
在研究非均相催化氧化体系时,反应途径的研究如同高层建筑的基础一样,是一项必不可少的首要任务。考虑到水净化稀溶液体系的特点,可采用多次循环累积表征和增加初始污染物浓度等策略进行碳平衡计算。对于产物和途径的鉴定,可以使用色谱分离和定量方法,结合质谱定性分析。在质谱检测中,应特别注意正负离子监测模式和电离效率(酸性/碱性流动相)等因素,以确保实验结果的可靠性。这些对于区分反应途径是否涉及矿化或聚合至关重要。
聚合途径不同于矿化途径,因为它们不会完全破坏有机污染物。如果聚合产物留在水中,它们可能继续构成污染风险。因此,合理开发聚合途径对未来净水技术的发展至关重要。基于聚合途径的DOTP技术具有广阔的应用前景。它不仅引起有机污染物间的聚合反应,而且使产物在固体表面形成厚层聚集。这种聚合途径和表面聚集行为的结合赋予DOTP特殊的净水潜力。与AOP相比,DOTP可以大大降低氧化剂/能量消耗,有助于减少CO2排放。然而,当有机物在固体表面积累和饱和时,最终会导致催化剂失活。为解决这一问题,DOTP的未来发展应着眼于降低催化剂成本和提高其积累量/能力。